Chapter 9 Mouse HSC (multiple technologies)
9.1 Introduction
The blood is probably the most well-studied tissue in the single-cell field, mostly because everything is already dissociated “for free”. Of particular interest has been the use of single-cell genomics to study cell fate decisions in haematopoeisis. Indeed, it was not long ago that dueling interpretations of haematopoeitic stem cell (HSC) datasets were a mainstay of single-cell conferences. Sadly, these times have mostly passed so we will instead entertain ourselves by combining a small number of these datasets into a single analysis.
9.2 Data loading
#--- data-loading ---#
library(scRNAseq)
sce.nest <- NestorowaHSCData()
#--- gene-annotation ---#
library(AnnotationHub)
ens.mm.v97 <- AnnotationHub()[["AH73905"]]
anno <- select(ens.mm.v97, keys=rownames(sce.nest),
keytype="GENEID", columns=c("SYMBOL", "SEQNAME"))
rowData(sce.nest) <- anno[match(rownames(sce.nest), anno$GENEID),]
#--- quality-control ---#
library(scater)
stats <- perCellQCMetrics(sce.nest)
qc <- quickPerCellQC(stats, percent_subsets="altexps_ERCC_percent")
sce.nest <- sce.nest[,!qc$discard]
#--- normalization ---#
library(scran)
set.seed(101000110)
clusters <- quickCluster(sce.nest)
sce.nest <- computeSumFactors(sce.nest, clusters=clusters)
sce.nest <- logNormCounts(sce.nest)
#--- variance-modelling ---#
set.seed(00010101)
dec.nest <- modelGeneVarWithSpikes(sce.nest, "ERCC")
top.nest <- getTopHVGs(dec.nest, prop=0.1)## class: SingleCellExperiment
## dim: 46078 1656
## metadata(0):
## assays(2): counts logcounts
## rownames(46078): ENSMUSG00000000001 ENSMUSG00000000003 ...
## ENSMUSG00000107391 ENSMUSG00000107392
## rowData names(3): GENEID SYMBOL SEQNAME
## colnames(1656): HSPC_025 HSPC_031 ... Prog_852 Prog_810
## colData names(3): cell.type FACS sizeFactor
## reducedDimNames(1): diffusion
## mainExpName: endogenous
## altExpNames(1): ERCCThe Grun dataset requires a little bit of subsetting and re-analysis to only consider the sorted HSCs.
#--- data-loading ---#
library(scRNAseq)
sce.grun.hsc <- GrunHSCData(ensembl=TRUE)
#--- gene-annotation ---#
library(AnnotationHub)
ens.mm.v97 <- AnnotationHub()[["AH73905"]]
anno <- select(ens.mm.v97, keys=rownames(sce.grun.hsc),
keytype="GENEID", columns=c("SYMBOL", "SEQNAME"))
rowData(sce.grun.hsc) <- anno[match(rownames(sce.grun.hsc), anno$GENEID),]
#--- quality-control ---#
library(scuttle)
stats <- perCellQCMetrics(sce.grun.hsc)
qc <- quickPerCellQC(stats, batch=sce.grun.hsc$protocol,
subset=grepl("sorted", sce.grun.hsc$protocol))
sce.grun.hsc <- sce.grun.hsc[,!qc$discard]library(scuttle)
sce.grun.hsc <- sce.grun.hsc[,sce.grun.hsc$protocol=="sorted hematopoietic stem cells"]
sce.grun.hsc <- logNormCounts(sce.grun.hsc)
set.seed(11001)
library(scran)
dec.grun.hsc <- modelGeneVarByPoisson(sce.grun.hsc) Finally, we will grab the Paul dataset, which we will also subset to only consider the unsorted myeloid population. This removes the various knockout conditions that just complicates matters.
#--- data-loading ---#
library(scRNAseq)
sce.paul <- PaulHSCData(ensembl=TRUE)
#--- gene-annotation ---#
library(AnnotationHub)
ens.mm.v97 <- AnnotationHub()[["AH73905"]]
anno <- select(ens.mm.v97, keys=rownames(sce.paul),
keytype="GENEID", columns=c("SYMBOL", "SEQNAME"))
rowData(sce.paul) <- anno[match(rownames(sce.paul), anno$GENEID),]
#--- quality-control ---#
library(scater)
stats <- perCellQCMetrics(sce.paul)
qc <- quickPerCellQC(stats, batch=sce.paul$Plate_ID)
# Detecting batches with unusually low threshold values.
lib.thresholds <- attr(qc$low_lib_size, "thresholds")["lower",]
nfeat.thresholds <- attr(qc$low_n_features, "thresholds")["lower",]
ignore <- union(names(lib.thresholds)[lib.thresholds < 100],
names(nfeat.thresholds)[nfeat.thresholds < 100])
# Repeating the QC using only the "high-quality" batches.
qc2 <- quickPerCellQC(stats, batch=sce.paul$Plate_ID,
subset=!sce.paul$Plate_ID %in% ignore)
sce.paul <- sce.paul[,!qc2$discard]9.3 Setting up the merge
common <- Reduce(intersect, list(rownames(sce.nest),
rownames(sce.grun.hsc), rownames(sce.paul)))
length(common)## [1] 17147Combining variances to obtain a single set of HVGs.
combined.dec <- combineVar(
dec.nest[common,],
dec.grun.hsc[common,],
dec.paul[common,]
)
hvgs <- getTopHVGs(combined.dec, n=5000)Adjusting for gross differences in sequencing depth.
9.4 Merging the datasets
We turn on auto.merge=TRUE to instruct fastMNN() to merge the batch that offers the largest number of MNNs.
This aims to perform the “easiest” merges first, i.e., between the most replicate-like batches,
before tackling merges between batches that have greater differences in their population composition.
Not too much variance lost inside each batch, hopefully. We also observe that the algorithm chose to merge the more diverse Nestorowa and Paul datasets before dealing with the HSC-only Grun dataset.
## DataFrame with 2 rows and 3 columns
## left right lost.var
## <List> <List> <matrix>
## 1 Paul Nestorowa 0.01069374:0.0000000:0.00739465
## 2 Paul,Nestorowa Grun 0.00562344:0.0178334:0.007026159.5 Combined analyses
The Grun dataset does not contribute to many clusters, consistent with a pure undifferentiated HSC population. Most of the other clusters contain contributions from the Nestorowa and Paul datasets, though some are unique to the Paul dataset. This may be due to incomplete correction though we tend to think that this are Paul-specific subpopulations, given that the Nestorowa dataset does not have similarly sized unique clusters that might represent their uncorrected counterparts.
library(bluster)
colLabels(merged) <- clusterRows(reducedDim(merged),
NNGraphParam(cluster.fun="louvain"))
table(Cluster=colLabels(merged), Batch=merged$batch)## Batch
## Cluster Grun Nestorowa Paul
## 1 54 646 228
## 2 109 372 46
## 3 0 38 189
## 4 0 157 523
## 5 0 216 472
## 6 128 67 339
## 7 0 6 29
## 8 0 135 211
## 9 0 19 0
## 10 0 0 386
## 11 0 0 382While I prefer \(t\)-SNE plots, we’ll switch to a UMAP plot to highlight some of the trajectory-like structure across clusters (Figure 9.1).
library(scater)
set.seed(101010101)
merged <- runUMAP(merged, dimred="corrected")
gridExtra::grid.arrange(
plotUMAP(merged, colour_by="label"),
plotUMAP(merged, colour_by="batch"),
ncol=2
)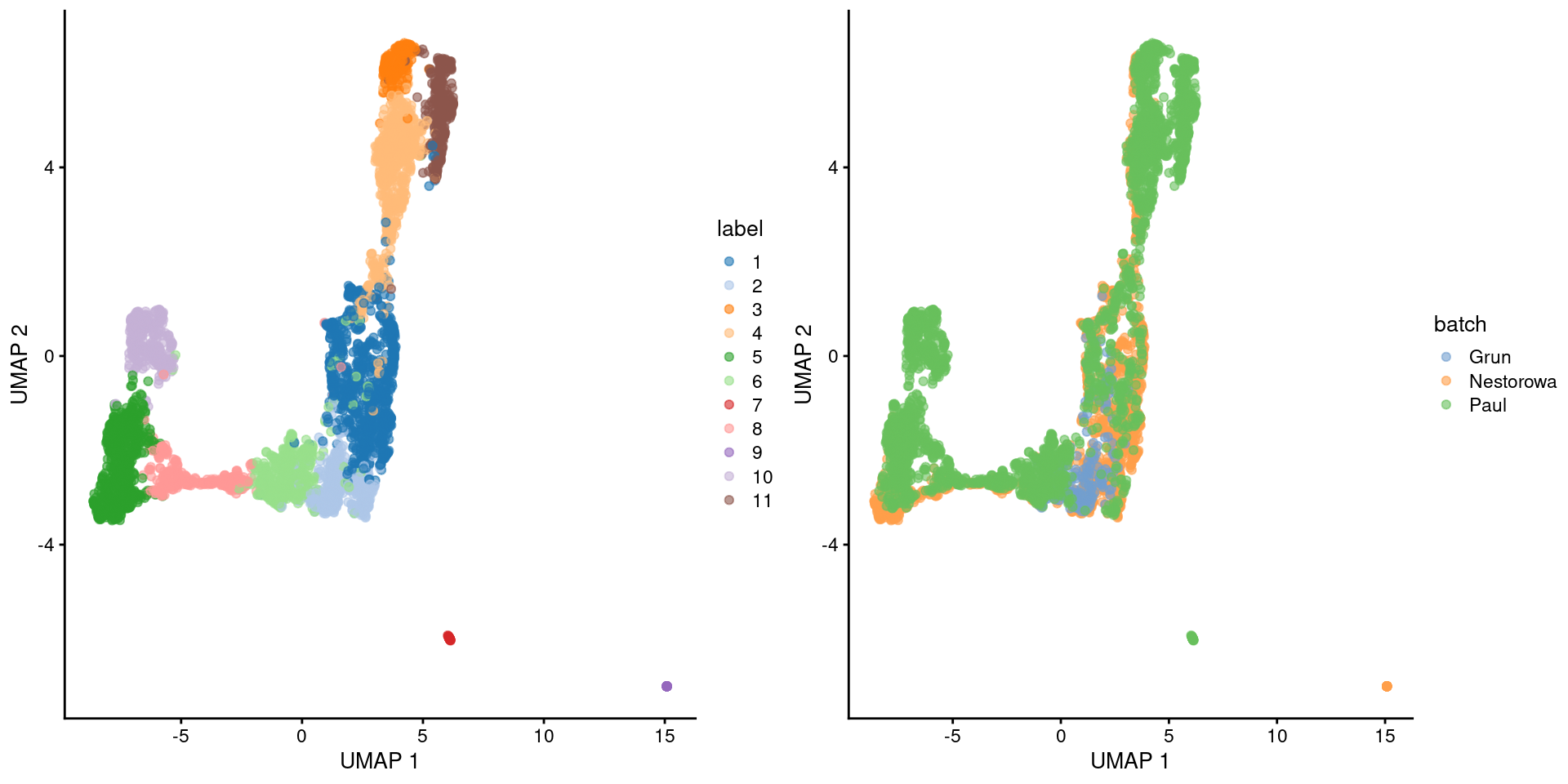
Figure 9.1: Obligatory UMAP plot of the merged HSC datasets, where each point represents a cell and is colored by the batch of origin (left) or its assigned cluster (right).
In fact, we might as well compute a trajectory right now. TSCAN constructs a reasonable minimum spanning tree but the path choices are somewhat incongruent with the UMAP coordinates (Figure 9.2). This is most likely due to the fact that TSCAN operates on cluster centroids, which is simple and efficient but does not consider the variance of cells within each cluster. It is entirely possible for two well-separated clusters to be closer than two adjacent clusters if the latter span a wider region of the coordinate space.
common.pseudo <- averagePseudotime(pseudo.out$ordering)
plotUMAP(merged, colour_by=I(common.pseudo),
text_by="label", text_colour="red") +
geom_line(data=pseudo.out$connected$UMAP,
mapping=aes(x=UMAP1, y=UMAP2, group=edge))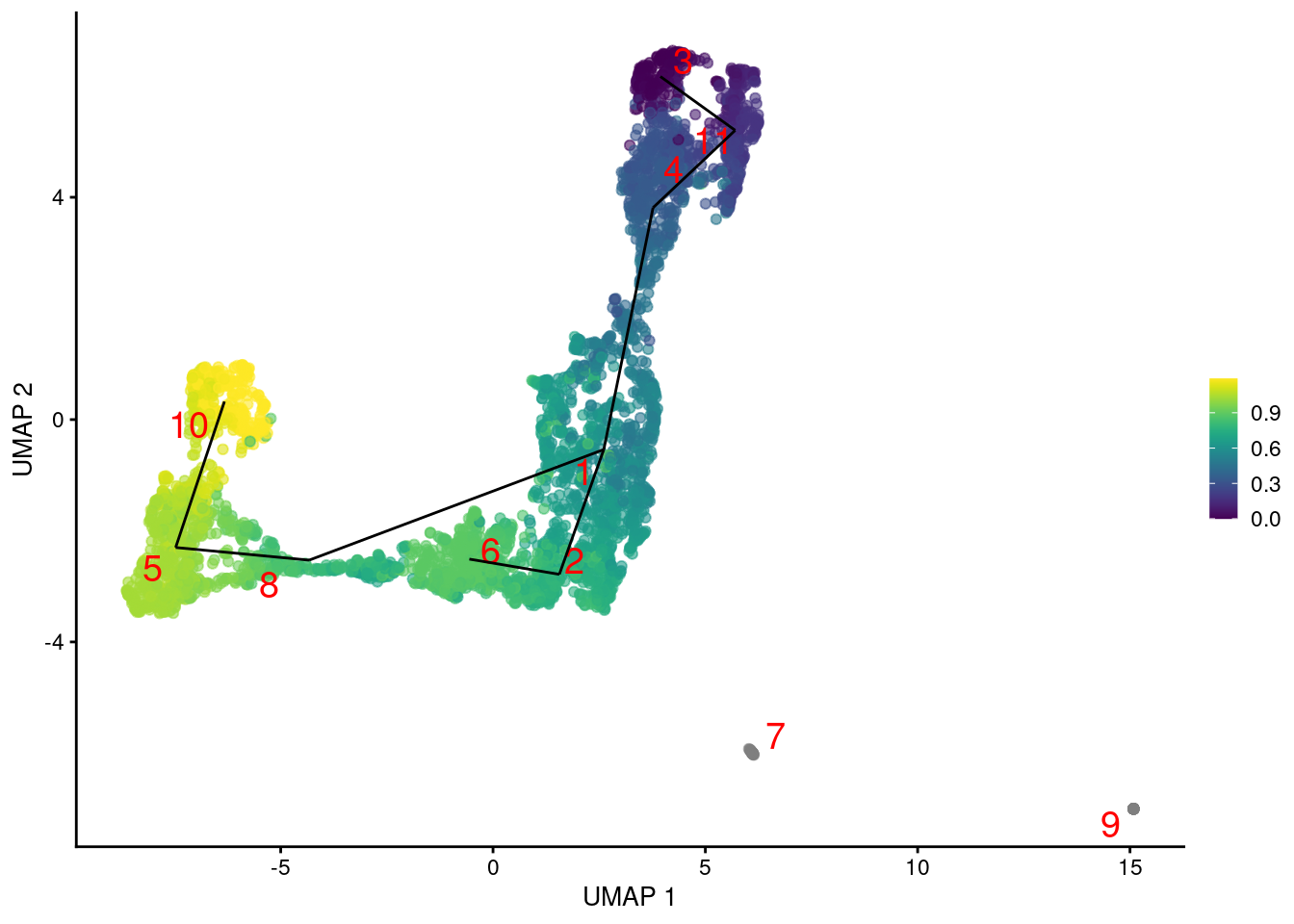
Figure 9.2: Another UMAP plot of the merged HSC datasets, where each point represents a cell and is colored by its TSCAN pseudotime. The lines correspond to the edges of the MST across cluster centers.
To fix this, we construct the minimum spanning tree using distances based on pairs of mutual nearest neighbors between clusters. This focuses on the closeness of the boundaries of each pair of clusters rather than their centroids, ensuring that adjacent clusters are connected even if their centroids are far apart. Doing so yields a trajectory that is more consistent with the visual connections on the UMAP plot (Figure 9.3).
pseudo.out2 <- quickPseudotime(merged, use.dimred="corrected",
dist.method="mnn", outgroup=TRUE)
common.pseudo2 <- averagePseudotime(pseudo.out2$ordering)
plotUMAP(merged, colour_by=I(common.pseudo2),
text_by="label", text_colour="red") +
geom_line(data=pseudo.out2$connected$UMAP,
mapping=aes(x=UMAP1, y=UMAP2, group=edge))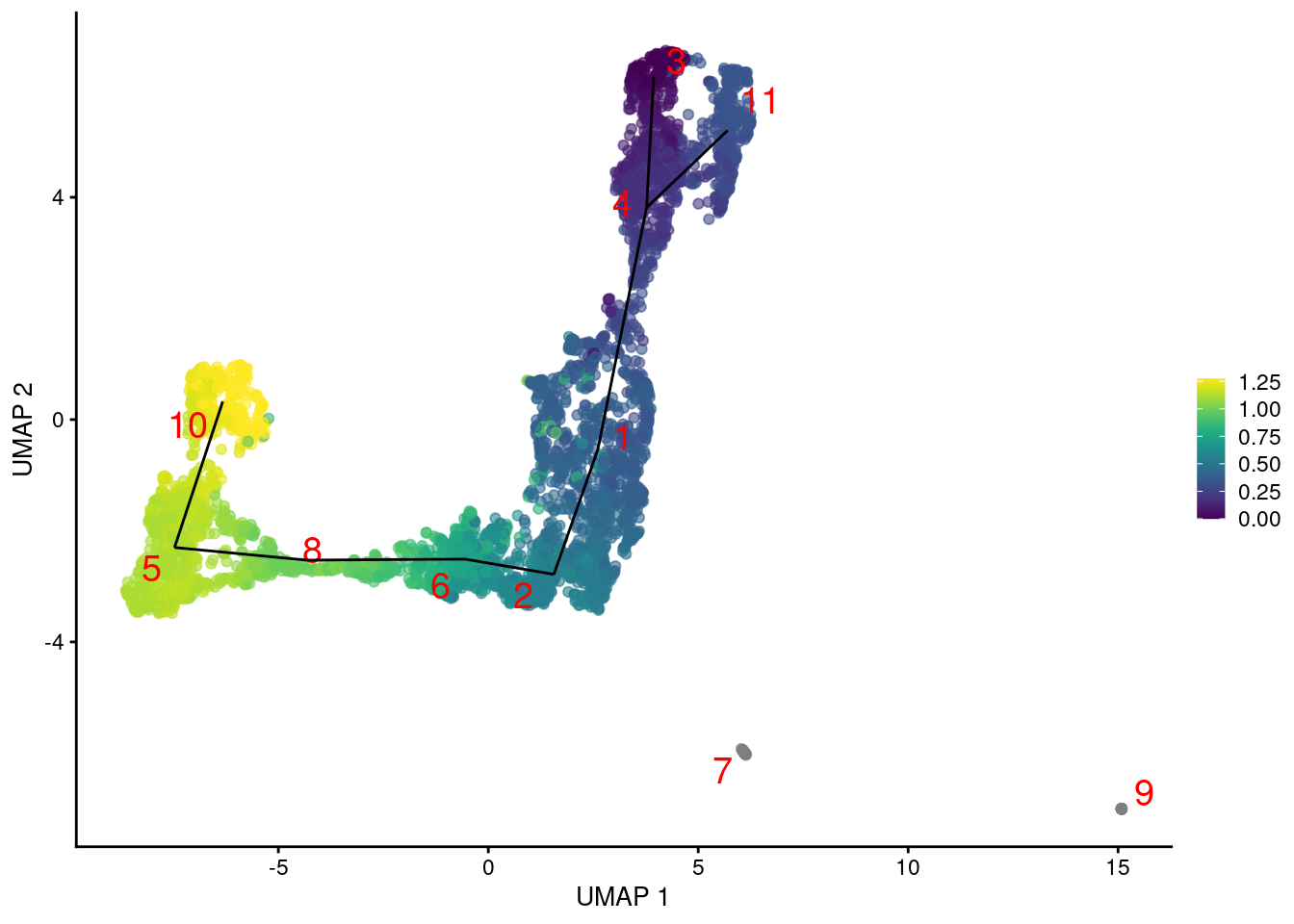
Figure 9.3: Yet another UMAP plot of the merged HSC datasets, where each point represents a cell and is colored by its TSCAN pseudotime. The lines correspond to the edges of the MST across cluster centers.
Session Info
R version 4.3.2 Patched (2023-11-13 r85521)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 22.04.3 LTS
Matrix products: default
BLAS: /home/biocbuild/bbs-3.18-bioc/R/lib/libRblas.so
LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.10.0
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_GB LC_COLLATE=C
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
time zone: America/New_York
tzcode source: system (glibc)
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] TSCAN_1.40.1 TrajectoryUtils_1.10.1
[3] scater_1.30.1 ggplot2_3.4.4
[5] bluster_1.12.0 batchelor_1.18.1
[7] scran_1.30.2 scuttle_1.12.0
[9] SingleCellExperiment_1.24.0 SummarizedExperiment_1.32.0
[11] Biobase_2.62.0 GenomicRanges_1.54.1
[13] GenomeInfoDb_1.38.6 IRanges_2.36.0
[15] S4Vectors_0.40.2 BiocGenerics_0.48.1
[17] MatrixGenerics_1.14.0 matrixStats_1.2.0
[19] BiocStyle_2.30.0 rebook_1.12.0
loaded via a namespace (and not attached):
[1] bitops_1.0-7 gridExtra_2.3
[3] CodeDepends_0.6.5 rlang_1.1.3
[5] magrittr_2.0.3 RcppAnnoy_0.0.22
[7] compiler_4.3.2 mgcv_1.9-1
[9] dir.expiry_1.10.0 DelayedMatrixStats_1.24.0
[11] vctrs_0.6.5 combinat_0.0-8
[13] pkgconfig_2.0.3 crayon_1.5.2
[15] fastmap_1.1.1 ellipsis_0.3.2
[17] XVector_0.42.0 labeling_0.4.3
[19] caTools_1.18.2 utf8_1.2.4
[21] promises_1.2.1 rmarkdown_2.25
[23] graph_1.80.0 ggbeeswarm_0.7.2
[25] xfun_0.42 zlibbioc_1.48.0
[27] cachem_1.0.8 beachmat_2.18.1
[29] jsonlite_1.8.8 later_1.3.2
[31] highr_0.10 DelayedArray_0.28.0
[33] BiocParallel_1.36.0 irlba_2.3.5.1
[35] parallel_4.3.2 cluster_2.1.6
[37] R6_2.5.1 bslib_0.6.1
[39] limma_3.58.1 jquerylib_0.1.4
[41] Rcpp_1.0.12 bookdown_0.37
[43] knitr_1.45 httpuv_1.6.14
[45] splines_4.3.2 Matrix_1.6-5
[47] igraph_2.0.1.1 tidyselect_1.2.0
[49] abind_1.4-5 yaml_2.3.8
[51] viridis_0.6.5 gplots_3.1.3.1
[53] codetools_0.2-19 plyr_1.8.9
[55] lattice_0.22-5 tibble_3.2.1
[57] shiny_1.8.0 withr_3.0.0
[59] evaluate_0.23 mclust_6.0.1
[61] pillar_1.9.0 BiocManager_1.30.22
[63] filelock_1.0.3 KernSmooth_2.23-22
[65] generics_0.1.3 RCurl_1.98-1.14
[67] fastICA_1.2-4 sparseMatrixStats_1.14.0
[69] munsell_0.5.0 scales_1.3.0
[71] xtable_1.8-4 gtools_3.9.5
[73] glue_1.7.0 metapod_1.10.1
[75] tools_4.3.2 BiocNeighbors_1.20.2
[77] ScaledMatrix_1.10.0 locfit_1.5-9.8
[79] XML_3.99-0.16.1 cowplot_1.1.3
[81] grid_4.3.2 edgeR_4.0.15
[83] colorspace_2.1-0 nlme_3.1-164
[85] GenomeInfoDbData_1.2.11 beeswarm_0.4.0
[87] BiocSingular_1.18.0 vipor_0.4.7
[89] cli_3.6.2 rsvd_1.0.5
[91] rappdirs_0.3.3 fansi_1.0.6
[93] S4Arrays_1.2.0 viridisLite_0.4.2
[95] dplyr_1.1.4 uwot_0.1.16
[97] ResidualMatrix_1.12.0 gtable_0.3.4
[99] sass_0.4.8 digest_0.6.34
[101] SparseArray_1.2.4 ggrepel_0.9.5
[103] dqrng_0.3.2 farver_2.1.1
[105] htmltools_0.5.7 lifecycle_1.0.4
[107] mime_0.12 statmod_1.5.0